paired end sequencing reads
One of the advantages of paired end sequencing over single end is that it doubles the amount of data. Paired-end tags PET sometimes Paired-End diTags or simply ditags are the short sequences at the 5 and 3 ends of a DNA fragment which are unique enough that they theoretically exist together only once in a genome therefore making the sequence of the DNA in between them available upon search if full-genome sequence data is available or upon further.

How Do You Put A Genome Back Together After Sequencing Genome Sequencing Learning Resources
In contrast RNA-seq on short RNAs 200 nt is typically carried out in single-end mode as the additional cost associated with paired-end would only translate into redundant sequence.

. This is typically about 30-40 bp in length and has. This aids in prediction of inversions deletions and. To create a paired end library random fragments of sequence are ligated to an adapter oligo.
Paired-end RNA sequencing RNA-seq is usually applied to the quantification of long transcripts such as messenger or long non-coding RNAs in which case overlapping pairs are discarded. All in forward direction. Illumina Paired End Sequencing.
The standard Illumina paired-end protocol produces reads oriented pointing toward each other just like good old fashioned Sanger paired reads but the insert size is much shorter. Paired-end sequencing facilitates detection of genomic rearrangements and repetitive sequence elements as well as gene fusions and novel transcripts. The 2 complementary DNA strands are oriented in opposite orientation and sequence reads from either end are generating results of those 2 different strands.
Read 1 and Read 2 would represent the ends of these fragments oriented inward. In general paired-end reads tend to be in a FR orientation have relatively small inserts 300 - 500 bp and are particularly useful for the sequencing of fragments that contain short repeat regions. Ad Gene Expression Profiling Chromosome Counting Epigenetic Changes Molecular Analysis.
Due to the way data is reported in these files special care has to be taken. The Illumina paired-end sequencing technology can generate reads from both ends of target DNA fragments which can subsequently be merged to increase the overall read length. Like everything there is the simple way and the correct way to do this.
An analysis by Whiteford et al. To start analysis of paired end Illumina sequence targeted amplicon data you need to create several files describing your data input and the raw sequences files which should be de-multiplexed on the Illumina barcodes already and are in a directory and gzipped. Both are methodologies that in addition to the sequence information give you information about the physical distance between the two reads in your genome.
These reads are assumed to be identical to the 100 5-most bases. Pairs come from the ends of the same DNA strand. This raw directory will not be modified in any way.
- Paired end gives an idea of the size of the insert and the diectionality of the mapping to the sequence assembly algorithms. Mate-pair fragments are generally in a RF conformation contain larger inserts 3 kb and enable sequence coverage of genomic regions containing large structural rearrangements. In paired-end reading it starts at one read finishes this direction at the specified read length and then starts another round of reading from the opposite end of the fragment.
Illumina gets sequence data from both strands of input sequence which means it outputs data from both ends of the input and is normally reported two files R1 and R2 often refereed to as mates files R1first mates R2second mates. Chaisson Brinza and Pevzner 2 recently determined that the paired read length threshold for de novo assembly of the E. This means your two reads are the reverse complement of the 100 3-most bases of the Watson strand and the Crick strand.
In genome sequencing projects one of the things we often need to do is split paired end sequence reads into the two ends. Paired end mate pair sequencing explanation biocc paired end or mate pair refers to how the library is made and then how it is sequenced. On sequencing using unpaired reads shows that ultra-short reads theoretically allow whole genome re-sequencing and de novo assembly of only small eukaryotic genomes.
Coli genome is 35 nt and 60 nt for the. There already exist tools for merging these paired-end reads when the target fragments are equally long. The first sequencing step is started by targeting SP1 to generate the forward read.
Paired-end reading improves the ability to identify the relative positions of various reads in the genome making it much more effective than single-end reading in resolving structural rearrangements such as. Imagine a 300bp DNA fragment with ends arbitrarily labeled A B. Fast and Accurate Next-Generation Sequencing Results Enabled by Ion Torrent Technology.
In conventional paired-end sequencing you simply sequence using the adapter for one end and then once youre done you start over sequencing using the adapter for the other end. Another supposed advantage is that it leads to more accurate reads because if say Read 1 see picture below maps to two different regions of the genome Read 2 can be used to help determine which one of the two regions makes more sense. Since paired-end reads are more likely to align to a reference the quality of the entire data set.
Paired-end sequencing allows users to sequence both ends of a fragment and generate high-quality alignable sequence data. Unlike two single fragment reads in paired-end sequence reads such as BAC-end sequences the two sequences in the pair have a known positional relationship in the original genome. The differences between PE and MP reads include.
Library preparation protocols -- In short PE protocols attach an adapter SP1 to the fwd end and another adapter SP2 to the reverse end. This provides an additional level of confidence over match scores and e-values in the accuracy of the positional assignment of the reads in the comparative genome.

Orthoclust An Orthology Based Network Framework For Clustering Data Across Multiple Species Rna Seq Blog Framework Macromolecules Data

Three Little Pigs Sequencing Three Little Pigs Little Pigs Three Little Pigs Story

New Publication From Csir Cimap Citing Genotypic S Ngs Services De Novo Sequencing And Comparative Analysis Of Holy And Sweet B Analysis Genome Sweet Basil

Illumina Doubles Output Of Benchtop Sequencer Miseq To 15 Gb What Is Epigenetics Exome Sequencing Epigenetics Doubles

An Iteration Normalization And Test Method For Differential Expression Analysis Of Rna Seq Data Rna Seq Blog Analysis Statistical Analysis Data

Deseq2 Moderated Estimation Of Fold Change And Dispersion For Rna Seq Data In Comparative High Throughput Sequencing Assa Molecular Biology Data Estimation
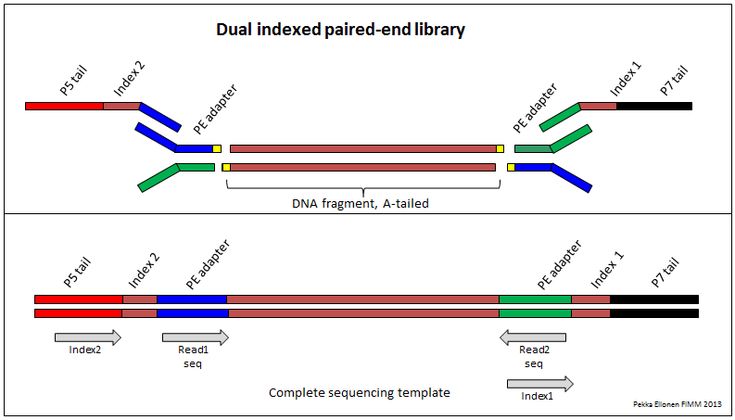
Demultiplexing Dual Indexed Miseq Fastq Files Seqanswers Data Science Hypothesis Index

The Advent Of Next Generation Sequencing And In Particular Rna Sequencing Rna Seq Technologies Has Expande Rna Sequencing Coding Next Generation Sequencing

Pin On Other Tools

Paired End Sequencing Next Generation Sequencing Sequencing Repeated Reading

Personal Genome Assembly Is A Critical Process When Studying Tumor Genomes And Other Highly Divergent Sequences The Data Visualization Reference Visualisation

Pin On Company Pipelines Pathways

In This Study Researchers From Stanford University Generated Two Paired End Rna Seq Datasets Of Differing Read Lengths 2 7 Rna Sequencing Long Reads Analysis

Read Build Write Mats Three Little Pigs Vocabulary Cards Vocabulary Cards Little Pigs Three Little Pigs
Seq2hla In Silico Hla Typing Using Standard Rna Seq Sequence Reads Reading Medical University Algorithm

Funpat Function Based Pattern Analysis On Rna Seq Time Series Data Dynamic Expression Data Nowadays Obtained U Analysis Functional Analysis Rna Sequencing

Pin On Services

Edgerun An R Package For Sensitive Functionally Relevant Differential Expression Discovery Using An Unco Next Generation Sequencing Expressions Data Science

Rna Extraction Method Read Length And Sequencing Layout Single End Versus Paired End Contribute Strongly To Var Interactive Notebooks Method Gene Expression